Публикации в СМИ
Дистрофия мышечная Дюшенна
Мышечная дистрофия Дюшенна — наследственная прогрессирующая мышечная дистрофия, характеризующаяся началом в раннем возрасте, симметричной атрофией мышц в сочетании с сердечно-сосудистыми, костно-суставными и психическими нарушениями, злокачественным течением; наследуется по рецессивному X-сцепленному типу. Вариант мышечной дистрофии Дюшенна — мышечная дистрофия Беккера — имеет более доброкачественное течение.
Генетические аспекты • Псевдогипертрофическая прогрессирующая мышечная дистрофия (мышечная дистрофия Дюшенна–Беккера, *310200, Xp21.2, ген DMD дистрофина, À рецессивное) — возникает в результате дефектов гена, кодирующего белок дистрофин • Дистрофин локализован в плазматической мембране скелетных мышечных волокон и кардиомиоцитов • Преобладающий пол — мужской, тем не менее мышечные дистрофии Дюшенна и Беккера могут встречаться у девочек при кариотипе X0, мозаицизмах X0/XX, X0/XXX и структурных аномалиях хромосом.
Патоморфология • Дистрофия мышечных волокон, первично-мышечный тип поражения • Фиброзные изменения в мышечных пучках • Местная воспалительная реакция.
Клиническая картина
• Мышечная дистрофия Дюшенна начинается в первые 1–3 года жизни обычно со слабости мышц тазового пояса.
• Уже на первом году жизни отмечают отставание в психомоторном развитии. Больные дети позднее начинают садиться, вставать, ходить.
• Постепенно развиваются слабость, патологическая мышечная утомляемость при физической нагрузке, изменение походки по типу утиной. Из горизонтального положения дети встают поэтапно с использованием рук (взбирание лесенкой).
• Отмечаются симметричные атрофии проксимальных групп мышц нижних конечностей (мышцы таза и бедра). Атрофия через 1–3 года распространяется на проксимальные группы мышц верхних конечностей.
• Атрофии мышц приводят к развитию лордоза, крыловидных лопаток, осиной талии.
• Характерна псевдогипертрофия икроножных мышц.
• Мышцы при пальпации плотные, безболезненные.
• Мышечный тонус обычно снижен в проксимальных группах мышц.
• Изменения рефлексов •• Коленные рефлексы исчезают на ранних стадиях заболевания •• Позднее исчезают рефлексы с двуглавой и трёхглавой мышц плеча •• Ахилловы рефлексы обычно длительное время остаются сохранными.
• Дистальная мускулатура конечностей поражается на поздних стадиях заболевания.
• Костно-суставные нарушения — деформации позвоночника, стоп, грудной клетки; рентгенологически обнаруживают сужение костномозгового канала, истончение коркового слоя диафизов длинных трубчатых костей.
• Сердечно-сосудистые расстройства — лабильность пульса, АД, приглушение тонов, расширение границ сердца, сердечная недостаточность, изменения на ЭКГ.
• Нейроэндокринные нарушения выявляют у 30–50% больных — синдром Иценко–Кушинга, адипозогенитальная дистрофия.
• Психические нарушения — олигофрения в форме дебильности или имбецильности.
• Клинические проявления мышечной дистрофии Беккера обычно начинаются в 10–15 лет. От мышечной дистрофии Дюшенна отличается доброкачественным течением и более поздним возникновением тяжёлых симптомов. Сухожильные рефлексы долгое время остаются сохранными. Поражения внутренних органов менее выражены, интеллект сохранён.
Лабораторные исследования. Для мышечной дистрофии Дюшенна типично раннее (с 5 дня жизни) увеличение активности КФК в крови (в 30–50 раз выше нормы).
Дифференциальная диагностика. Мышечную дистрофию Дюшенна–Беккера дифференцируют от других мышечных дистрофий, рахита, врождённого вывиха бедра.
ЛЕЧЕНИЕ
Режим амбулаторный с наблюдением у невропатолога, хирурга-ортопеда, терапевта и профпатолога, работника социальной сферы и протезиста.
Мероприятия • Лечение мышечной дистрофии Дюшенна направлено на поддержании физической активности пациента и улучшение качества его жизни; как правило, быстро становится неэффективным • Физические упражнения выполняют систематически и по определённой схеме. Короткие перерывы показаны при возникновении болей в мышцах и мышечной усталости • Использование протезов позволяет больным двигаться и замедляет формирование сколиоза • Поддержание дыхания, ИВЛ во время сна для предотвращения синдрома ночной гиповентиляции • Экспериментальные методы, в особенности генная терапия (гены дистрофина и утрофина), чрезвычайно перспективны, хотя и не получили пока клинического распространения.
Оперативное лечение. Ортопедическое вмешательство необходимо при наличии контрактур и фиксации суставов.
Лекарственная терапия • ГК (преднизолон по 0,75 мг/кг/сут) увеличивают мышечную силу у мальчиков, страдающих мышечной дистрофией Дюшенна, замедляя прогрессирование заболевания • При длительной стероидной терапии необходим тщательный контроль развития побочных эффектов, включающий наблюдение за массой тела, АД, состоянием слизистой оболочки ЖКТ и иммунной системы.
Наблюдение. Ранняя диагностика поражения внутренних органов позволяет увеличить продолжительность жизни пациентов.
Профилактика состоит в генетическом консультировании.
Синонимы • Прогрессирующая мышечная дистрофия Дюшенна • Псевдогипертрофическая мышечная дистрофия Дюшенна • Дистрофия Дюшенна • Болезнь Дюшенна • Миопатия псевдогипертрофическая • Миопатия псевдогипертрофическая Дюшенна.
МКБ-10 • G71.0 Мышечная дистрофия • M62.5 Истощение и атрофия мышц, не классифицированные в других рубриках • M62.8 Другие уточнённые поражения мышц
Примечания • Термин «псевдогипертрофическая прогрессирующая мышечная дистрофия» объединяет мышечные дистрофии Дюшенна и Беккера • Мышечная дистрофия Дюшенна описана в 1853 г. Дюшенном • Мышечная дистрофия Беккера описана в 1955 г. Беккером.
Мышечная дистрофия Дюшенна
Впервые псевдогипертрофическая прогрессирующая мышечная дистрофия была описана французским неврологом в середине девятнадцатого века — Гийомом Дюшенном, благодаря которому и получило свое название – дистрофия Дюшенна. Это одно из самых частых среди наследственных мышечно-дистрофических заболеваний, встречается с частотой 3-4 человека на сто тысяч населения.
Благодаря своей яркой клинической картине и достаточно широкой распространенности, многие люди впервые сталкиваясь с заболеванием (а также, к сожалению, часто и некоторые из врачей) считают, что есть только этот вид мышечной дистрофии, однако их множество и есть некоторые тонкости в симптоматике и диагностике заболевания. Об этом и пойдет речь в данной статье.
Информация для врачей. Все варианты наследственных мышечных дистрофий по МКБ10 шифруются в рубрике G71.0. Указывается тип заболевания по автору (может указываться также дифференциальный ряд, например, напоминающая дистрофию Дюшенна или Беккера). Также указывается стадия, скорость прогрессирования, степень слабости определенных групп мышц в баллах.
Причины
Заболевание наследственное, сцепленное с X-хромосомой, поэтому болеют практически всегда мальчики. Девочки являются носителем патологического гена (мальчики редко доживают до половозрелого возраста, к тому же, как правило, стерильны). В хромосоме происходит изменение структуры гена, отвечающего за синтез белка дистрофина.
Хоть содержание дистрофина в скелетной мускулатуре предельно мало (тысячные доли процента), без него быстро развивается некроз мышечной ткани, развивается прогрессирующая дистрофия мышц. Если ген повреждается на участке, полностью разрушающем синтез белка-дистрофина, развивается дистрофия Дюшенна. При вовлечении в процесс малозначимых отделов белка, заболевание принимает форму дистрофии Беккера.
Симптомы
Начало развития симптомов начинается в раннем детском возрасте, чаще от 1 до 3 лет. Изначально отмечается отставание в моторном развитии, ребенок поздно начинает ходить, часто спотыкается при ходьбе, быстро устает. Позже развивается постоянная патологическая утомляемость мышц. Ребенок практически не может взбираться по лестнице. Походка начинает напоминать «утиную».
Характерным симптомом является симптом «лесенки»: попытка встать из положения сидя происходит с использованием рук, постепенно, медленно, в несколько этапов.
Постепенно начинает отмечаться атрофия мышц, вначале проксимальных отделов нижних, потом верхних конечностей. Позже атрофируются мышцы тазового пояса, бедер, спины, плечевого пояса. Почти всегда развивается «осиная» талия, искривление позвоночника, выпирание лопаток (крыловидные лопатки).
 Практически всегда имеет место характерный симптом прогрессирующей мышечной дистрофии Дюшенна – псевдогипертрофия мышц голеней. Мышцы, хоть и увеличены в объеме, они не имеют достаточной силы, очень болезненны на ощупь.
Практически всегда имеет место характерный симптом прогрессирующей мышечной дистрофии Дюшенна – псевдогипертрофия мышц голеней. Мышцы, хоть и увеличены в объеме, они не имеют достаточной силы, очень болезненны на ощупь.
Можно выделить три стадии заболевания:
— I стадия – слабость проявляется лишь при значимой физической нагрузке (обычно первый год течения болезни).
— II стадия – затруднен подъем по лестнице, быстро развивается слабость при ходьбе.
— III стадия – представляет собой параличи, контрактуры мышц с невозможностью cсамостоятельного передвижения.
По видам течения подразделяется на:
Быстрое прогрессирование. Способность к передвижению утрачивается быстро, в течение первых 4-5 лет с начала заболевания.
Средний темп прогрессирования: передвигаться пациент не может спустя 10 лет.
Медленное прогрессирование: выраженных двигательных нарушений через 10 лет от начала болезни нет. Обычно такой вариант характерен для других типов мышечных дистрофий, нежели дистрофии Дюшенна.
Диагностика
Клиническая картина очень яркая. Часто заболевание ставится после выяснения генетического анамнеза (наличие случаев в семье), неврологического осмотра. В неврологическом статусе отмечается пропадание коленных рефлексов, чуть позже исчезают рефлексы с бицепса, трицепса. Ахилловы рефлексы долгое время сохранны.
Внешне может выявиться деформация суставов стопы, имеются признаки кардиомиопатии: нарушение пульса, глухость сердечных тонов, расширение полостей сердца по ЭхоКГ, изменения на электрокардиограмме.
Важным фактором является повышение биохимических показателей креатинфосфокиназы (фермент-показатель распада мышц). Активность данного фермента увеличивается в десятки раз. Имеется прямая корреляция между степенью увеличения активности фермента и выраженностью проявлений дистрофии Дюшенна. [!] В сложных диагностических ситуациях проводят цитологическое исследование.
Лечение и прогноз жизни
Лечение симптоматическое. Используются гормональные препараты для остановки разрушения мышечного волокна, фосфолипиды в качестве защиты клеток мышц от разрушения, элементы лечебной гимнастики. Внедряются в практику различные ортопедические приспособления для облегчения передвижения. Массаж строго противопоказан в большинстве случаев, так как может приводить к ускорению распада мышц. Лечение наследственных заболеваний – дело будущего.
Прогноз жизни для пациентов неблагоприятный. Течение заболевание прогрессирующее. Неизбежен летальный исход. Как правило, к семилетнему возрасту развивается выраженная симптоматика, приводящая к 13-14 годам к полной обездвиженности. Больные редко доживают до 18-20 лет.

Автор сайта: Алексей Борисов — практикующий невролог, отоневролог (специалист по вопросам головокружения).
— Окончил Иркутский государственный медицинский университет.
— Заведую кабинетом головокружения.
— Регулярно прохожу курсы повышения квалификации, участвую и выступаю с докладами на образовательных конференциях, в том числе с международным участием.
— Имею большое количество печатных научных публикаций.
Прогрессирующая мышечная дистрофия Дюшенна

Прогрессирующая мышечная дистрофия Дюшенна — наследуемая сцеплено с Х-хромосомой патология мышечной системы, проявляющаяся в первые 3-5 лет жизни и характеризующаяся быстро распространяющейся и усугубляющейся мышечной слабостью. Первоначально поражаются мышцы тазового пояса и бедер, затем — плеч и спины, постепенно наступает обездвиженность. Миодистрофия сопровождается скелетными деформациями и поражением сердца. Диагностика дистрофии Дюшенна включает неврологическое и кардиологическое обследование, определение уровня КФК, электромиографию, консультацию генетика, ДНК-анализ, биопсию мышц. Лечение симптоматическое. В связи со слабостью дыхательной мускулатуры на заключительном этапе заболевания требуется ИВЛ.
Общие сведения
Причины мышечной дистрофии Дюшенна
Развитие мышечной дистрофии Дюшенна связано с наличием мутации в 21-ом локусе короткого плеча Х-хромосомы в гене, кодирующем белок дистрофин. Около 70% случаев болезни вызваны дефектным геном дистрофина, полученным от матери — носительницы патологической мутации. Остальные 30% связаны с появлением свежих мутаций в яйцеклетках матери. В отличие от миодистрофии Беккера, при дистрофии Дюшенна генетические аберрации приводят к сдвигу рамки считывания ДНК и полному прекращению синтеза дистрофина, что и обуславливает более тяжелое течение патологии.
В норме входящий в сарколемму миоцитов дистрофин обеспечивает ее целостность и устойчивость к растяжению, возникающему при сократительной активности мышечных волокон. Отсутствие дистрофина влечет за собой нарушение целостности сарколеммы, разрушение миоцитов и их замещение жировой и соединительной тканью. Клинически этот процесс выражается прогрессирующим снижением способности мышц к сокращению, утратой мышечной силы и тонуса, атрофией мышц.
Симптомы мышечной дистрофии Дюшенна
Дебют миодистрофии Дюшенна приходится на период от 1 до 5 лет. Как правило, уже на 1-ом году жизни заметно некоторое отставание моторного развития ребенка. Отмечается задержка сроков начала сидения, самостоятельного вставания и ходьбы. Когда ребенок начинает ходить, он отличается неуклюжестью и большей, по сравнению со сверстниками, неустойчивостью; часто спотыкается.
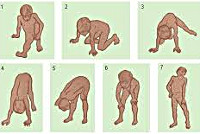
Мышечная слабость возникает на 3-4-ом годах жизни. Первоначально она выражается в патологически повышенной утомляемости при ходьбе по лестнице или на длинные расстояния. Со временем становится заметной типичная для миодистрофий утиная походка. Обращают на себя внимание особенности поведения ребенка — каждый раз, поднимаясь из положения сидя на корточках, он активно опирается руками о собственное тело, как бы взбираясь по нему как по лесенке (симптом Говерса).
Мышечные атрофии начинаются с мышц бедер и тазового пояса. Для дистрофии Дюшенна характерно их быстрое восходящее распространение на плечевой пояс, мускулатуру спины и проксимальных отделов рук. Вследствие мышечных атрофий формируется «осиная» талия и отстоящие от спины «крыловидные» лопатки. Типичным симптомом выступает псевдогипертрофия икроножных мышц. Наблюдается выпадение сухожильных рефлексов: вначале — коленных, затем — рефлексов с трицепса и бицепса плеча. Ахилловы и карпорадиальные рефлексы могут длительное время быть сохранны. Со временем развиваются ретракции сухожилий и мышечные контрактуры.
Прогрессирующая мышечная дистрофия Дюшенна сопровождается нарушениями в костно-суставной системе. Характерны искривление позвоночника (кифоз, усиленный лордоз, сколиоз), деформации грудной клетки (килевидная или седловидная), деформации стоп. Сердечно-сосудистые расстройства обусловлены развитием кардиомиопатии и включают аритмию, лабильность артериального давления, глухость тонов сердца. У 50% больных фиксируются нейроэндокринные расстройства — адипозогенитальная дистрофия, синдром Иценко-Кушинга и др. Около 30% больных страдает олигофренией, как правило, ограничивающейся степенью дебильности. Могут отмечаться СДВГ, расстройства по типу аутизма, дислексия, нарушения краткосрочной памяти.
Уже к 7-10-летнему возрасту дистрофия Дюшенна приводит к выраженным двигательным ограничениям. К 12 годам больные, как правило, утрачивают способность ходить, а к возрасту 15 лет большинство пациентов полностью теряют возможность самостоятельных движений. Распространение дистрофического процесса на дыхательную мускулатуру приводит к прогрессирующему падению жизненной емкости легких (ЖЕЛ) и, в конечном итоге, невозможности совершать дыхательные движения.
Диагностика мышечной дистрофии Дюшенна
Установить диагноз миодистрофии Дюшенна помогает анамнез, неврологическое обследование, результаты электрофизиологического тестирования, определение креатинфосфокиназы (КФК) в биохимическом анализе крови, морфологическое и иммунохимическое исследование образцов мышечной ткани, генетическое консультирование и анализ ДНК. При этом дифференциальную диагностику следует проводить с другими миопатиями — метаболической, воспалительной, миодистрофией Беккера, мышечной дистрофией Дрейфуса, дистрофией Эрба-Рота, а также с полиневропатиями, полимиозитом, БАС.
Электронейро- и электромиография определяют сохранность проведения импульсов по нервным волокнам, пониженную амплитуду М-ответа, что свидетельствует о первично-мышечном типе поражения. Характерным является 30-50-кратный подъем уровня креатинфосфокиназы. На консультации генетика проводится генеалогическое исследование, позволяющее выявить наличие случаев миодистрофии Дюшенна в семье больного и определить женщин, являющихся носительницами мутантного гена дистрофина. Диагностика ДНК позволяет выявить аномалии в гене дистрофина. Следует учитывать, что невыявление мутации при ДНК-анализе не говорит о ее отсутствии, поскольку поиск точковых мутаций обычно не входит в задачи анализа из-за его большой длительности и трудоемкости.
В случаях, когда имеется клиническая картина миодистрофии, а анализ ДНК не выявил наличие мутации, показана биопсия мышц. Морфологическое исследование биоптата определяет разнокалиберность и некроз миоцитов, их замещение соединительнотканными элементами. Иммунохимический анализ говорит о полном отсутствии дистрофина в исследуемых мышечных волокнах.
Дополнительно осуществляется обследование костно-мышечной и сердечно-сосудистой систем — проводится консультация ортопеда, рентгенография позвоночника, обзорная рентгенография ОГК, консультация кардиолога, ЭКГ, эхокардиография. По показаниям рекомендуется консультация эндокринолога, пульмонолога и др. специалистов.
Лечение мышечной дистрофии Дюшенна
Терапия, применяемая в клинической практике, пока включает лишь необходимые симптоматические мероприятия. Для улучшения метаболизма мышечной ткани возможно назначение анаболических стероидов (метандиенона, нандролона деканоата), АТФ, актопротекторов (этилтиобензимидазола); для облегчения нервно-мышечной передачи — неостигмина. С целью минимизировать образование контрактур и продлить двигательную активность пациентов проводится ЛФК, массаж, физиотерапия.
Важное значение имеет контроль дыхательной функции и газового состава крови. При падении ЖЕЛ до 40% рекомендована искусственная вентиляция легких в период сна. В дальнейшем время ИВЛ растет пропорционально снижению ЖЕЛ. В начале ИВЛ может осуществляться при помощи масочного аппарата. Затем необходима трахеостомия, и ИВЛ проводится путем присоединения аппарата к трахеостомической трубке. Современные портативные аппараты ИВЛ работают на батареях и могут быть закреплены на инвалидной коляске.
Поиск эффективных способов лечения дистрофии Дюшенна — задача, над решением которой трудятся сегодня специалисты в области неврологии, биохимии, генной инженерии. Из перспективных разработок в этом направлении можно выделить лечение стволовыми клетками, активацию гена утрофина, являющегося наиболее близким аналогом дистрофина, технологию пропуска экзонов.
Прогноз и профилактика
Профилактические мероприятия направлены на выявление женщин-носительниц аномального гена дистрофина и предупреждение рождения у них больного ребенка. В рамках профилактических мер проводятся консультации генетика для планирующих беременность супружеских пар, консультации беременных и пренатальная ДНК-диагностика.
Дистрофия Дюшена

Весь контент iLive проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.
Заболевание, которое называется дистрофия Дюшена, связывают с повреждением генных структур, ответственных за продукцию крупного мышечного белка дистрофина. Такая патология передается наследственным путем, аутосомно-рецессивным типом наследования: то есть, данная патология как бы прячется, либо проявляется через поколение. Данный тип сцеплен с X-хромосомой.
 [1]
[1]
Код по МКБ-10
Причины дистрофии Дюшена
Патология появляется вследствие генной мутации, имеющей место в области хр21. Более четверти таких патологий связывают со стойким изменением генотипа в материнской яйцеклетке. Остальные случаи объясняют гетерозиготностью мамы пациента по патологии мутагенеза в гене дистрофина.
Принято считать, что примерно 7% всех периодически возникающих случаев заболевания – это следствие образования в женском яичнике нескольких клеточных генераций с мутированными и обычными аллеями дистрофина. При этом наиболее распространенным типом мутации (около 65%) являются значительные потери участков хромосомы. У 5% пациентов обнаруживают удвоение участка хромосомы, а в оставшихся случаях патологии – point mutation, когда затрагивается один или несколько нуклеотидов, тогда как к мутации относятся более протяженные дефекты гена.
Патология передается по аутосомно-рецессивному типу, со сцепкой с X-хромосомой (поражает мужские особи). Больше половины таких патологий возникают спонтанно, в связи с мутацией генов.
При генетическом обследовании большую роль играет определение у сестер пациента скрытых признаков заболевания. От такой носительницы мутированного гена может произойти передача патологии 50% её детей мужского пола, а 50% её дочек станут такими же носителями мутированного гена.
Женщины, которые обладают поврежденным геном, делятся им со своим ребенком, хотя сами миопатией не страдают. В основном заболеванию подвержены мальчики. Девочки также могут заболеть, но это случается крайне редко: такое может произойти лишь при дефекте структуры хромосом.
[2]
Симптомы дистрофии Дюшена
Начальные симптомы дистрофии Дюшена можно заметить уже в возрасте от 1 до 5 лет. Для больного ребенка характерно торможение ранней двигательной активности. При попытках ходить самостоятельно (у детей, старше 1 года) можно наблюдать постоянные падения, запутывание ножек, быструю усталость. Если малыш все-таки начинает ходить, то при этом он переваливается с ноги на ногу (утиная походка), ему сложно подниматься по ступенькам и вставать с колен.
Постепенно у маленьких пациентов возникает увеличение объемов различных мышечных групп, что внешне похоже на сильно накачанные мышцы. С дальнейшим течением и усугублением патологии такое увеличение переходит, наоборот, в уменьшение мышц.
Заболевание распространяется по телу восходящим путем: от мышц ног и малого таза до спины, плеч и рук.
Уже на начальных этапах заболевания можно наблюдать понижение сухожильных рефлексов. Далее развивается искривление позвоночника, грудная клетка становится седло- или килевидной, деформируются стопы. Возникают проблемы с сердечной мышцей: появляются признаки нарушения сердечного ритма и левожелудочковая гипертрофия. У четверти пациентов обнаруживают симптомы умственной отсталости: чаще всего это проявляется признаками олигофрении.
Приблизительно к 12-летнему возрасту пациенты перестают ходить, через 2-3 года полностью теряют способность совершать движения. В 20-30 лет большее количество таких больных умирает. На поздних этапах заболевания мышечная слабость переходит и на дыхательную и глотательную систему. Летальный исход наступает от присоединившихся бактериальных инфекций или от недостатка дыхательной и сердечной деятельности.
[3], [4], [5], [6]
Формы
[7], [8], [9], [10]
Мышечная дистрофия Дюшена
Мышечная дистрофия Дюшена, к счастью, встречается относительно редко и проявляется в мышечной слабости. Согласно статистике, такая патология встречается приблизительно у одного малыша из 3000 новорожденных детей. Кроме того, известны несколько достаточно редких форм миопатии, отличающиеся менее выраженными проявлениями.
Развитие мышечной дистрофии связано с медленным разрушением соединений между нервными и мышечными волокнами.
Девочки, родившиеся от матери с поврежденным геном, также могут стать носителем такого повреждения, хотя заболевание у них практически никогда не проявляется.
Кроме дистрофии Дюшена, медицина выделяет ещё и другие виды миопатий, встречающиеся крайне редко:
[11], [12], [13], [14], [15]
Прогрессирующая дистрофия Дюшена
Прогрессирующая дистрофия Дюшена – это серьезная, наиболее распространенная форма миопатии. Развивается она в младенческом возрасте, обычно у детей до 3-х лет, изредка в более старшем возрасте. Практически у всех пациентов (за небольшим исключением) наблюдается увеличение размеров икроножных мышц (иногда в совокупности с дельтовидными и четырехглавыми мышцами). Такое увеличение часто связано с жировой инфильтрацией мышц, однако в некоторых случаях в действительности увеличиваются именно мышцы.
Уменьшение объема мышечной массы наблюдается в основном в области спины и тазового пояса. Наряду с атрофическими расстройствами зачастую можно отмечать наличие умственной отсталости.
Не редкостью являются нарушения целостности и формы костей даже вследствие незначительных нагрузок и травм. Спустя 5-10 лет от первых проявлений патологии можно обнаружить поражение сердечной мышцы, что выражается тахикардией и нарушениями ЭКГ. Характерным признаком считается усиленная активность сывороточной креатинкиназы.
В целом, заболевание протекает тяжелее, чем при остальных формах миопатии. Атрофические изменения быстро распространяются по всему организму. Большая часть пациентов к 10-летнему возрасту практически лишена возможности двигаться. Такие больные крайне редко способны дожить до 30 лет, погибая от сопутствующих заболеваний.
Диагностика дистрофии Дюшена
Диагноз дистрофии Дюшена должен быть подтвержден методом генетического тестирования, однако в некоторых случаях возможно назначение других исследований.
Очень важно определить точный диагноз, если специалисты подозревают у ребенка миопатию. Причем сделать это необходимо в самом скором времени. Квалифицированное лечение доктор назначит на основании этих исследований, предварительно проведя беседу с родителями и разъяснив им все особенности заболевания.
[16], [17], [18], [19]
Как обследовать?
К кому обратиться?
Лечение дистрофии Дюшена
В настоящее время лекарства от дистрофии Дюшена ещё не изобрели. Хотя научные исследования на эту тему проводятся достаточно интенсивно: над этим работают ученые Великобритании, Израиля и США. Новейшие методы, находящиеся в стадии разработки:
На данный момент специалисты предлагают следующее лечение дистрофии Дюшена:
«Эликсира исцеления» от дистрофии Дюшена, к сожалению, не существует, поэтому при поисках эффективного лечения с крайней осторожностью относитесь к препаратам и процедурам, которые могут быть представлены вашему вниманию как «панацея».
Не покупайте неизвестное лекарство, если нет достоверных подтверждений его эффективности. Помните, что вы можете потратить большую финансовую сумму, и, к тому же, не только не помочь, но и навредить своему малышу.
Профилактика
Конечно, говорить о профилактике наследственного заболевания, связанного с мутацией гена, сложно. Безусловно, в семьях, в которых имеются случаи рождения детей с мышечной дистрофией Дюшена, в обязательном порядке необходимо проводить консультации генетика, желательно ещё до начала планирования беременности молодой парой.
Если ребенок с патологией мышц уже появился на свет, то необходимо приложить все усилия, чтобы не допустить потерю мышечной ткани и как можно дольше поддерживать у ребенка двигательную активность. В таких ситуациях недопустимо опускать руки и позволять мышцам атрофироваться. Следует заниматься с малышом специальной гимнастикой, чтобы сохранить амплитуду движений в суставах. Для предупреждения контрактур рекомендуется применять поддерживающие корсеты и фиксаторы.
Дети, которые хоть немного, но могут ходить, должны это делать как можно чаще. Не загоняйте ребенка в постель: пусть будет активным, это позволит мышцам замедлить регрессию. Таким детям необходимо уделять максимум внимания и заботы, они не должны чувствовать себя обделенными или обиженными.
Хороший эффект дает плавание, которым заниматься можно и даже нужно. Помните: ничегонеделание (постоянный постельный режим) лишь ускорит прогрессирование болезни. Необходимо позволять больному, по крайней мере, самому удовлетворять свои потребности.
[20], [21], [22], [23], [24], [25]