Болезнь Штаргардта
Болезнь Штаргардта – наследственное заболевание сетчатки, которое проявляется дистрофическими изменениями ее макулярной зоны и приводит к потере центрального зрения. Дебют заболевания приходится на детский или юношеский возраст. У пациентов отмечаются центральные скотомы и нарушения цветового зрения. Прогрессирование болезни Штаргардта приводит к полной слепоте. Диагностика проводится с помощью офтальмоскопии, флуоресцентной ангиографии и ЭФИ сетчатки. Для лечения применяется инъекционная терапия (витамины, антиоксиданты, ангиопротекторы), физиотерапия, проводятся реваскуляризирующие операции, разрабатывается методика аутологичной тканевой терапии.
Общие сведения
В 1997 году генетики обнаружили мутацию гена АВСR, вызывающую нарушение выработки белка, который должен переносить энергию фоторецепторным клеткам. Неполноценность переносчика АТФ приводит к гибели фоторецепторов сетчатки глаз. Различные виды наследственно обусловленной макулярной дистрофии встречаются в 50% случаев патологии глаз. Из них болезнь Штаргардта составляет около 7%. Нозологическая форма диагностируется с частотой 1:10000 и характеризуется прогрессирующим течением. Двусторонняя патология глаз начинается в молодом возрасте (от 6 до 21 года) и приводит к тяжелым последствиям, вплоть до полной потери зрения. Заболевание имеет социальную значимость, потому что приводит к инвалидности в молодом возрасте.
Причины развития болезни Штаргардта
Наследование не зависит от половой принадлежности пациента и родителей. Патология передается преимущественно по аутосомно-рецессивному типу, то есть наследование патологии не связано с полом (аутосомное – связано с неполовыми хромосомами) и не всегда передается будущему поколению (рецессивный путь наследования). По последним данным врачей-генетиков, патология гена может передаваться и по доминантному типу. При доминантном типе наследования дефектов гена – контролера синтеза белка-транспортера АТФ – заболевание протекает легче и нечасто приводит к инвалидности. Большинство рецепторных клеток макулы (верхушки) желтого пятна глазного дна функционируют. У пациентов с доминантным типом наследования болезнь протекает с минимумом проявлений. Больные сохраняют работоспособность и могут даже водить автотранспорт.
Основная причина дегенерации клеток макулы заключается в том, что они страдают от дефицита энергии. Дефект гена приводит к синтезу неполноценного белка, транспортирующего молекулы АТФ через мембрану клеток желтого пятна – центра сетчатки глаза, в котором фокусируется графическое и цветное изображение. В области желтого пятна нет кровеносных сосудов. Питание клеток-колбочек осуществляется за счет белков-переносчиков АТФ из близлежащей сосудистой оболочки (хориоидеи). Белки переносят через мембрану внутрь клеток-колбочек молекулы АТФ.
В нормальных условиях родопсин фоторецепторов поглощает фотон света, трансформируясь в транс-ретиналь и опсин. Затем транс-ретиналь под действием энергии АТФ, которую приносят белки-переносчики, превращается в ретиналь, который соединяется с опсином. Так восстанавливается родопсин. При наследственной мутации гена образуется неполноценный белок-переносчик. В результате нарушается восстановление родопсина и скапливается транс-ретиналь. Он превращается в липофусцин и оказывает прямое токсическое действие на клетки-колбочки.
Классификация болезни Штаргардта
Перицентральная форма характеризуется появлением скотомы в стороне от точки фиксации. Человек способен фокусировать взгляд, но отмечает выпадения в одной из сторон от центра поля зрения в виде полумесяца. Со временем скотома приобретает вид темного кольца. Центропериферическая форма начинается с центра и стремительно распространяется к периферии. Темное пятно разрастается и полностью перекрывает поле зрения.
Симптомы болезни Штаргардта
Проявления заболевания начинаются в возрасте 6-7 лет. У всех пациентов, независимо от типа наследования, наблюдаются центральные скотомы. При благоприятном течении скотомы относительные: пациент видит яркие предметы с четкими контурами и не различает объекты со слабой цветовой гаммой. У многих больных отмечается нарушение цветового зрения по типу красно-зеленой дисхромазии, при которой человек видит светло-зеленый цвет как темно-красный. В то же время, некоторые пациенты не отмечают изменения восприятия цветной гаммы.
В начальной фазе заболевания не изменяются границы периферического зрения, при прогрессировании центральные скотомы расширяются, что приводит к полной слепоте. Одновременно с появлением выпадения центрального зрения уменьшается его острота. В завершающей стадии болезни Штаргардта зрительный нерв атрофируется. Человек полностью теряет зрение. Не наблюдается изменений других органов, как в начальной, так и в терминальной стадии заболевания.
Диагностика болезни Штаргардта
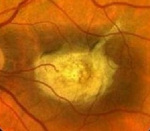
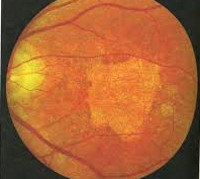
Заболевание начинается в детском возрасте – это один из главных признаков для дифференциальной диагностики. С помощью офтальмоскопии обнаруживается широкое кольцо пониженной пигментации, которое окружает темный центр. Вокруг бледного кольца отмечается следующее кольцо гиперпигментированных клеток. Картина напоминает «бычий глаз» или «кованую бронзу». Фовеолярный рефлекс отрицательный. Макулярное возвышение не определяется. При осмотре желтого пятна отмечаются желтовато-белые пятна разной величины и конфигурации. Со временем границы включений размываются, пятна приобретают серый оттенок либо полностью исчезают.
Во время проведения периметрии при болезни Штангардта отмечаются положительные или отрицательные (пациент их не ощущает) центральные скотомы. При центральной форме заболевания развивается красно-зеленая дейтеранопия. Для периферической формы не характерно нарушение цветового восприятия. Пространственная контрастная чувствительность изменяется по всему диапазону: отсутствует в области высоких частот (в центральном участке до 6-10 градусов) и снижается в области средних частот.
В начальной стадии заболевания отмечается снижение показателей макулярной электрографии при центральной форме дистрофии. При дальнейшем прогрессировании электрические потенциалы не регистрируются. При расположении дистрофии по средней периферической зоне в начальной стадии отмечается нормальная электрография и электроокулография. Затем значения колбочковых и палочковых компонентов элетроретинографии снижаются до субнормальрных. Заболевание протекает бессимптомно – без нарушения остроты зрения и восприятия цвета. Границы поля зрения находятся в пределах нормы. Незначительно снижена темновая адаптация.
С помощью флуоресцентной ангиографии на фоне «бычьего глаза» не обнаруживаются зоны гипофлуоресценции, просматриваются капилляры, «молчащая» или «темная» сосудистая оболочка. В зонах атрофии заметны гиперфлуоресцирующие участки клеток пигментного эпителия сетчатки. Гистологическое исследование в центральной зоне глазного дна определяет повышенное количество пигмента – липофусцина. Отмечается комбинация гипертрофированных и атрофированных клеток пигментного эпителия.
Молекулярно-генетический анализ позволяет заметить мутацию гена до начала проявлений болезни. Чтобы обнаружить замену нуклеотидов, проводится ПЦР в режиме реального времени при использовании нескольких ДНК-зондов – «молекулярных маяков». Дифференциальная диагностика болезни Штаргардта осуществляется с приобретенными лекарственными дистрофиями, пятнами сетчатки Кандори, семейными друзами, ювенильным ретиношизисом, доминантной прогрессирующей фовеальной, колбочковой, колбочно-палочковой и палочко-колбочковой дистрофией.
Лечение и прогноз болезни Штаргардта
Этиологического лечения нет. В качестве общего вспомогательного лечения применяются парабульбарные инъекций таурина и антиоксидантов, введение сосудорасширяющих средств (пентоксифиллин, никотиновая кислота), стероидных препаратов. Проводится витаминотерапия для укрепления сосудов и улучшения кровоснабжения (вит. группы В, А, С, Е). Показаны физиотерапевтические методы лечения: лекарственный электрофорез, ультразвук, лазерстимуляция сетчатки. Применяется методика реваскуляризации сетчатки путем трансплантации в зону желтого пятна пучка мышечных волокон. Разрабатывается патогенетическая регенерационная офтальмологическая технология аутологичной тканевой терапии с помощью стволовых клеток жировой ткани пациента.
Болезнь Штаргардта начинается в раннем возрасте и быстро приводит к инвалидности по зрению. В редких случаях, при доминантном типе наследования, зрение падает медленно. Пациентам рекомендуется наблюдение офтальмолога, прием витаминных комплексов и ношение солнцезащитных очков.
Болезнь Штаргардта является наследственной патологией сетчатки глаза, сопровождающейся дегенеративными изменениями в ее центральной (макулярной) зоне, что влечет за собой утерю центрального зрения. Как правило, заболевание проявляет себя еще в детском или юношеском возрасте центральными скотомами и нарушением цветовосприятия. При прогрессировании болезни Штаргардта, исходом процесса становится полная слепота. В диагностике патологии, применяют офтальмоскопию, флуоресцентную ангиографию и ЭФИ сетчатки. В качестве лечения, назначается инъекционная терапия препаратами витаминов, ангиопротекторов, антиоксидантов, а также физиотерапия. Кроме того, проводятся реваскуляризирующие операции и применяется, находящаяся в разработке, методика аутологичной тканевой терапии.

Рис.1 Центральная (макулярная) зона сетчатки, которая поражается при болезни
О патологии
Болезнь Штаргардта носит и другое официалное название – ювенильная макулярная дегенерация. Именно оно наиболее полно отражает суть заболевания, которое начинается в раннем (ювенильном) возрасте. Патология характеризуется поражением области макулы сетчатки – рецепторного аппарата анализатора зрения. Второе свое название болезнь получила по имени, описавшего ее в начале ХХ века, немецкого ученого Карла Штаргардта. Патология описывалась, как врожденное поражение сетчатки в макулярной области, которое было характерно для представителей одного рода. Офтальмоскопические признаки заболевания полиморфны и типичны, это: атрофия хориоидеи, «битая (кованая) бронза», «бычий глаз». Патогенетически, заболевание обозначается, как «желтопятнистая абиотрофия сетчатки», что отражает визуализируемые изменения на глазном дне.
Болезнь вызывает мутация гена АВСR с нарушением синтеза белка, отвечающего за транспорт энергии клеткам-фоторецепторам. Это было выяснено учеными-генетиками в 1997 году. Тогда же удалось доказать, что патология переносчика АТФ становится причиной гибели фоторецепторов сетчатой оболочки.
Наследственно обусловленная макулярной дегенерация – заболевание довольно частое, ее различные виды встречаются практически в половине случаев патологии глаз. Болезнь Штаргардта, при этом, составляет примерно 7% всех случаев. Частота нозологической формы болезни, характеризующейся прогрессирующим течением, составляет 1:10000 в популяции.
Патология имеет двусторонний характер течения, дебютирует в молодом возрасте (до 21 года) и очень часто становится причиной полной потери зрения. Социальная значимость болезни Штаргардта весьма велика, так как она приводит к инвалидности людей молодого трудоспособного возраста.
Причины возникновения
Наследование патологии происходит вне зависимости от половой принадлежности детей и родителей. Преимущественно она передается аутосомно-рецессивным путем, то есть поражает потомков родителей-носителей заболевания, однако не в 100% случаев. Последние исследования врачей-генетиков выявили, что патология гена способна передаваться также и по доминантному пути.
В случае доминантного пути передачи болезни, она протекает легче и к инвалидности приводит довольно редко. Большинство клеток- рецепторов в макуле (не верхушке желтого пятна) остаются функциональными, поэтому патология имеет минимум проявлений. Люди сохраняют работоспособность, острота зрения позволяет даже водить автотранспорт.
Родопсин фоторецепторов в нормальных условиях поглощает фотоны света, с превращением в опсин и транс-ретиналь. Затем, под действием энергии АТФ, приносимой белками-переносчиками, транс-ретиналь превращается в ретиналь и соединяется с опсином. Это процесс восстановления родопсина. Наследственная мутация гена становится причиной синтеза неполноценного белка-переносчика. В итоге восстановление родопсина нарушается, происходит скопление транс-ретиналя. Он трансформируется в липофусцин, который и оказывает токсическое воздействие напрямую на клетки-колбочки.
Типы и виды болезни Штаргардта
Видовая изменчивость патологии объясняется распространенностью зоны поражения макулярной области. Специалисты классифицируют болезнь Штаргардта по следующим формам:
Центральная форма характеризуется поражением клеток в центре области макулы. Процесс сопровождается выпадением центрального зрения. Человек отмечает появление центральной скотомы (темного пятна перед глазом). Происходит выпадение центральной зоны из поля зрения и видимое изображение в точке фиксации взгляда получается с темным пятном.
Перицентральная форма сопровождается возникновением в стороне от точки фиксации скотомы. Способность фокусировать взгляд остается, но с одной из сторон от центра поля зрения отмечает его выпадение в форме полумесяца. С прогрессом заболевания, скотома принимает вид темного кольца.
Для центрально-периферической формы характерно стремительное ухудшение зрения от центра к периферии. Постепенно разрастаясь, темное пятно полностью закрывает поле зрения.

Рис.2 Важность посещения врача-офтальмолога: чем раньше выявлена болезнь, тем выше шанс сохранить зрение
Признаки заболевания
Начало заболевания приходится на возраст 6-7 лет, тогда же возникают первые его симптомы. Вне зависимости от типа наследования, все пациенты отмечают возникновение центральных скотом. Ели, течение заболевания благоприятное, скотомы относительные. Те есть, человек способен четко видеть яркие предметы, но объекты, имеющие слабую окраску, различает хуже. Многие больные страдают нарушением цветовосприятия по типу красно-зеленой дисхромазии, когда человек воспринимает зеленый цвет, как красный. Правда, в некоторых случаях пациенты изменения цветового восприятия могут не отмечать.
В начальной стадии болезни, границы периферического зрения не изменены, но при прогрессировании центрально скотомы, они сужаются, что ведет к слепоте.
Одновременно с выпадением центрального зрения снижается и его острота. На итоговой фазе болезни Штаргардта происходит атрофия зрительного нерва и человек теряет зрение полностью. Изменений прочих глазных сред, на всех стадиях, не наблюдается.
Диагностика
Начало заболевания характерно для детского возраста. Это и является одним из главных диагностических его признаков.
При выполнении офтальмоскопии, выявляется широкое кольцо локализованное на сетчатке с пониженной пигментацией. Оно окружает темный центр здоровой ткани. Вокруг бледного кольца имеется следующее кольцо клеток с гиперпигментацией. Офтальмологи определяют такую картину, как «бычий глаз», «кованая бронза». Макулярное возвышение не определяется, нет фовеолярного рефлекса. При осмотре области макулы определяются бело-желтые пятна разной конфигурации и размера. Границы включений имеют тенденцию размываться со временем, пятна становятся сероватыми либо исчезают полностью.
Проведение периметрии при болезни Штангардта выявляет отрицательные (неощутимые) и положительные центральные скотомы. Определяется красно-зеленая дейтеранопия, если патологический процесс протекает в центральной форме, и отсутствие нарушения цветового восприятия в периферической форме. Изменение пространственной контрастной чувствительности по всему диапазону: ее отсутствие в зоне высоких частот (в центральном участке до 6-10 градусов), снижается в зоне средних частот.
Показатели макулярной электрографии в начальной стадии заболевания при центральной форме дистрофии снижены. Электрические потенциалы при дальнейшем прогрессировании процесса, не регистрируются. Если дистрофии расположена по средней периферической зоне, нормальная электрография регистрируется только в начальной стадии. Затем при электроретинографии, значения компонентов колбочек и палочек становятся субнормальными. Болезнь протекает без симптомов: нарушения остроты зрения и цветовосприятия не наблюдается. В норме границы поля зрения. Темновая адаптация снижена незначительно.

Лечение и прогноз
Как такового лечения, направленного на устранение заболевания нет. В качестве общей терапии показано внутриглазное введение таурина и антиоксидантов, расширяющих сосуды средств (никотиновая кислота, пентоксифиллин), гормональных препаратов. Также назначается витаминотерапия (вит. группы В, А, С, Е), направленная на укрепление сосудов, чтобы улучшить кровоснабжение и питание пораженной зоны сетчатки, ретинопротекторы (ретиналамин). Целесообразно проведение физиотерапии: ультразвук, лекарственный электрофорез, лазерстимуляция.
В стадии разработки находится метод реваскуляризации сетчатки посредством пересадки в макулярную зону пучка мышечных волокон. Кроме того, на этапе исследования находится технология патогенетической регенерационной аутологичной тканевой терапии с использованием стволовых клеток жировой ткани пациента.
Для болезни Штаргардта характерно начало в детском возрасте и быстрый прогресс до состояния инвалидизации пациента по зрению. Зрение ухудшается медленно только в очень редких случаях доминантного пути наследования заболевания. В качестве профилактики, рекомендуется прием витаминных комплексов, длительное наблюдение офтальмолога, применение солнцезащитных очков.
Пациенты с болезнью Штаргардта нуждаются в динамическом наблюдении врача-ретинолога. Специалисты нашей клиники – признанные лидеры лечения заболеваний сетчатки. Применение инновационных технологий, новейшая аппаратура от лучших мировых производителей и индивидуальный подход к каждому пациенту, обеспечивают гарантированное получение максимально высоких результатов даже в самых сложных случаях.
Наследственные ретинальные дистрофии
Рубрика МКБ-10: H35.5
Содержание
Определение и общие сведения [ править ]
Синонимы: Ювенильная макулярная дегенерация
Выделяют четыре формы болезни Штаргардта в зависимости от локализации патологического процесса: в макулярной области, на средней периферии (fundus flavimaculatus), в парацентральной области, а также смешанную форму при локализации в центре и на периферии.
Этиология и патогенез [ править ]
В ПЭС происходит интенсивное накопление липофусцина. Он ослабляет окислительную функцию лизосом, увеличивает рН клеток ПЭС, что приводит к нарушению их мембранной целостности.
Клинические проявления [ править ]
При центральной форме дистрофии Штаргардта по мере развития процесса офтальмоскопическая картина макулярной области имеет различный вид: от «битого металла» до «бычьего глаза», «кованой бронзы» и атрофии хороидеи.
Феномен «бычий глаз» офтальмоскопически виден как тёмный центр, окружённый широким кольцом гипопигментации, за которым обычно следует другое кольцо гиперпигментации. Сосуды сетчатки не изменены, ДЗН (диск зрительного нерва) бледен с темпоральной стороны, что связано с атрофией нервных волокон в папилломакулярном пучке. Фовеолярный рефлекс и макулярное возвышение (umbo) отсутствуют.
Наследственные ретинальные дистрофии: Диагностика [ править ]
Время возникновения заболевания (в детском или юношеском возрасте) может играть важную роль в его диагностике.
Гистологически отмечают увеличение количества пигмента в центральной зоне глазного дна, атрофию прилежащего ПЭС, комбинацию атрофии и гипертрофии пигментного эпителия. Жёлтые пятна представлены липофусциноподобным материалом.
При периметрии у всех больных с болезнью Штаргардта обнаруживают относительные или абсолютные центральные скотомы разной величины в зависимости от сроков и распространения процесса с раннего детского или юношеского возраста. При жёлто-пятнистом глазном дне изменений в макулярной области не отмечают, поле зрения может быть не изменено.
ЭРГ и ЭОГ. Макулярная ЭРГ снижается уже в начальных стадиях центральной формы дистрофии Штаргардта и не регистрируется в развитых стадиях.
В начальных стадиях fundus flavimaculatus ганцфельд ЭРГ и ЭОГ остаются в пределах нормы; в развитых стадиях снижаются колбочковые и палочковые компоненты ЭРГ, которая становится субнормальной, так же изменяются показатели ЭОГ. У больных при этой форме симптомы отсутствуют. Острота зрения, цветовое зрение, поле зрения находятся в пределах нормы. Темновая адаптация может быть нормальной или незначительно сниженной.
На ФАГ при типичном феномене «бычьего глаза» на нормальном фоне обнаруживают зоны «отсутствия», или гипофлюоресценции, с видимыми хориокапиллярами, «тёмную», или «молчащую», хороидею. Отсутствие флюоресценции в макулярной области объясняют накоплением липофусцина, экранирующего флуоресцеина. Участки с гипофлюоресценцией могут становиться гиперфлюоресцирующими, что соответствует зоне атрофии ПЭС.
Дифференциальный диагноз [ править ]
Сходство клинической картины различных дистрофических заболеваний макулярной области затрудняет диагностику. Дифференциальный диагноз болезни Штаргардта следует проводить с семейными друзами, fundus albipunctatus, пятнами сетчатки Кандори, доминантной прогрессирующей фовеальной дистрофией, колбочковой, колбочко-палочковой и палочко-колбочковой дистрофией, ювенильным ретиношизисом, вителлиформной макулярной дистрофией, приобретёнными лекарственными дистрофиями (например, хлороквиновой ретинопатией).
Наследственные ретинальные дистрофии: Лечение [ править ]
Патогенетически обоснованного лечения нет.
Информация для пациента (краткие рекомендации)
Рекомендуют ношение солнцезащитных очков в качестве профилактической меры от повреждающего действия света, применение общеукрепляющей витаминотерапии.
Острота зрения снижается прогрессивно, особенно быстро в детском и юношеском возрасте, что зависит от размера выраженности изменений в макулярной области.
Профилактика [ править ]
Прочее [ править ]
Синонимы: вителлиформная макулярная дистрофия Беста.
В северной Швеции и Дании частота возникновения патологии оценивется на уровне 1/5000 и 1/67000 соответственно. Заболеванию больше подвержены мужчины, чем женщины (3:1).
Болезнь Беста наследуется аутосомно-доминантно с полной пенетрантностью.
Течение болезни бессимптомное. Болезнь Беста обнаруживают случайно при осмотре детей в возрасте 5-15 лет. Изредка пациенты предъявляют жалобы на затуманивание зрения, затруднения при чтении текстов с мелким шрифтом, метаморфопсии. Острота зрения варьирует в зависимости от стадии болезни от 0,02 до 1,0. Изменения в большинстве случаев асимметричные, двусторонние.
В зависимости от изменений в макулярной области, определяемых офтальмо-скопически, выделяют пять стадий заболевания, хотя развитие болезни не всегда проходит через все стадии.
• Стадия 1 (превителлиформная). Характеризуется минимальными нарушениями пигментации в виде мелких жёлтых очажков в макуле.
• Стадия 2 (вителлиформная). Классическая вителлиформная киста в макуле.
• Стадия 3. Разрыв кисты и различные фазы резорбции её содержимого.
• Стадия 4. Резорбция содержимого кисты,
• Стадия 5. Формирование фиброглиального рубца с субретинальной неоваскуляризацией или без неё.
Диагноз у пациентов с болезнью Беста устанавливают на основании результатов офтальмоскопии, ФАГ, ЭРГ и ЭОГ. В трудных случаях в диагностике может помочь обследование других членов семьи.
Пренатальная диагностика и преимплантационная генетическая диагностика возможны для семей, в которых известны мутации, вызывающие заболевание.
Лечение является симптоматическим и включает использование средств коррекции зрения для лиц со значительным его снижением. Рекомендовано ежегодное офтальмологическое обследование для лиц всех возрастов. Следует избегать курения, так как оно увеличивает риск неоваскулярной макулярной дегенерации. Для купирования субфовеальной хориоидальной неоваскуляризации могут быть использованы фотодинамическая терапия с использованием вертепорфина, непосредственно лазерная фотокоагуляция или анти-VEGF препараты (бевацизумаб). Также может быть использована транскорнеальная электрическая ретинальная стимуляция.
Ухудшение зрительных функций по мере развития и резорбции желточной кисты.
Вителлиформная макулярная дистрофия взрослых
Мутация обнаружена в гене периферина (RDS). В отличие от болезни Беста, патологии в фовеолярной области при вителлиформной макулярной дистрофии взрослых развиваются в зрелом возрасте, затрагивают меньшие участки и не прогрессируют.
ЭОГ, как правило, не изменена.
Развивающееся в раннем детском возрасте наследственное заболевание, характеризующееся дистрофическими изменениями пигментного эпителия и фоторецепторов и имеющее типичную картину глазного дна с пигментными очагами, схожими с пигментными костными тельцами.
а) Генетические типы: аутосомно-доминантный, аутосомно-рецессивный, сцепленный с Х-хромосомой.
б) Клинические формы: типичная (пигментный ретинит); атипичная (беспигментная форма пигментного ретинита, секторальный подковообразный пигментный ретинит, инвертированный (центральная форма); белоточечный пигментный ретинит (retinitis punctata albescens).
Этиология и патогенез
Более 3000 мутации в более чем 57 различных генов или локусов в настоящее время известно.
Большинство мутаций, приводящих к дегенерации сетчатки, происходит в генах, кодирующих синтез белков каскада фототрансдукции и обмена ацетата ретинола (витамина А) (родопсина, α- и β-субъединиц циклического гуанозинмонофосфата, арестина, АТФ-связывающего кассетного переносчика ретиналя, G-протеина, клеточного ретинальдегида, связывающего протеин, и гена RPGR-трансдукции и др.), а также в генах, ответственных за фактор транскрипции.
Различают аллельную и неаллельную разновидности пигментного ретинита.
В связи с поражением палочковой системы возникает ночная слепота (отсутствие ночного зрения, никталопия). Темновая адаптация нарушена уже в начальной стадии заболевания, порог световой чувствительности повышен как в палочковой, так и в колбочковой части.
К другим формам пигментного ретинита относят: пигментный ретинит инвертированный, пигментный ретинит без пигмента, белоточечный пигментный ретинит (retinitis punctata albescens) и псевдопигментный ретинит. Каждая из этих форм имеет характерную офтальмоскопическую картину и ЭРГ-симптоматику.
Лечение в первую очередь направлено на замедление прогрессирования заболевания. Витамин А и лютеин используется в качестве антиоксидантов. Ацетазоламид внутрь или местно дорзоламид используются для уменьшения кистозного отека макулы. Экстракция хрусталика требуется, если катаракта снижает остроту зрения. Солнцезащитные очки с фильтрацией коротких волн улучшает зрение.
Определение и общие сведения
Синдром Бера включает в себя атрофию зрительного нерва и сопутствующие неврологические симтомы. Синдром Бера клинически похож на синдром Костеффа, но без метаболических нарушений.
Синдром Бера наследственное заболевание, передается аутосомно-рецессивно. Распространненость неизвестна, оба пола в равной степени подвержены заболеванию.
Атрофия зрительного нерва приводит к нарушению остроты зрения от умеренной до тяжелой степени тяжести, которое проявляется в раннем возрасте. Нарушение остроты зрения часто сопровождается сенсорным нистагмом.
Неврологические симтомы имеют внутри- и межсемейную изменчивость. Они могут включать в себя миоклоническую эпилепсию, прогрессирующую спастическую параплегию вследствие участия пирамидного тракта, дизартрию, экстрапирамидные симптомы, атаксию, недержание мочи, умственную отсталость, потерю чувствительности вследствие поражения задних столбов спинного мозга и мышечные контрактуры (в основном в нижних конечностях). Эти неврологические симптомы проявляются постепенно в детстве и становятся заметны во втором десятилетии. К этому возрасту большинство больных с синдромом Бера передвигаются только при помощи ходунков.
Часто пациентам требуется хирургическая коррекция контрактур ахиллова сухожилия и большой приводящей мышцы бедра.
Синонимы: синдром пигментный ретинит-тугоухость, синдром Ашера
Определение и общие сведения
Синдром Ушера характеризуется сочетанием нейросенсорной глухоты (обычно врожденной) с пигментным ретинитом и прогрессирующей потерей зрения.
Этиология и патогенез